Mutational Signatures (v3.6 - May 2026)
CN9 · GRCh37 · COSMIC v104
Mutational profile

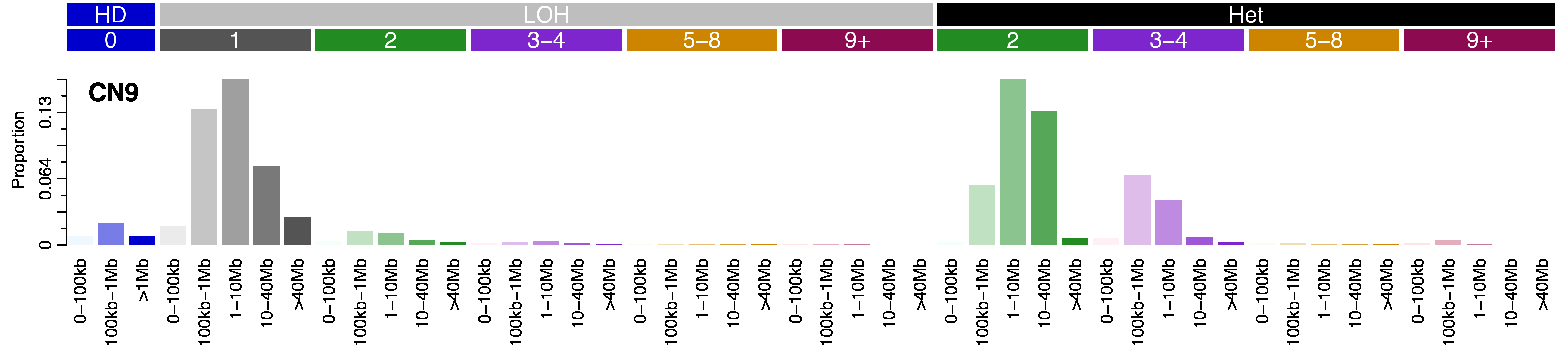
Copy number signatures are defined by a 48 context copy number classification scheme. The scheme incorporates loss-of-heterozygosity status, total copy number state, and segment length to categorise segments from allele-specific copy number profiles.
The scheme corresponds to three heterozygosity states:
- heterozygous segments with copy number of [A>0, B>0] (numbers reflect the counts for major allele A and minor allele B)
- segments with LOH with copy number of [A>0, B=0]
- segments with homozygous deletions [A=0, B=0]
Segments are further subclassified into 6 classes based on the sum of major and minor allele (total copy number) and chosen for biological relevance:
- total copy number (TCN)=0 (homozygous deletion)
- TCN=1 (deletion leading to LOH)
- TCN=2 (wild type, including copy-neutral LOH)
- TCN=3 or 4 (minor gain)
- TCN=5 to 8 (moderate gain)
- TCN>=9 (high-level amplification).
Each of the heterozygous and LOH total copy number states are then organised into five classes based on the size of their segments: 0 – 100kb, 100kb – 1Mb, 1Mb – 10Mb, 10Mb – 40Mb, and >40Mb (the largest category for homozygous deletions was restricted to >1Mb) in order to capture focal, large scale, and chromosomal-scale copy number changes. This gives a total of 48 copy number context to categorise segments into (3x homozygous deletion categories, 5*5=25x LOH categories, 4*5=20 heterozygous categories).
Proposed aetiology
CN9 is a signature of chromosomal instability on a diploid background. Evidence for this comes from both simulations of chromosomal instability and simulations of chromothripsis (N.B. that there is no current signature of ‘canonical chromothripsis’; this is difficult to distinguish from the effects of regular chromosomal instability with the current classification system). In addition, CN9 is associated with non-genome-doubled samples.
Notes
CN9 is associated with both deletion and duplication rearrangement classes (see Hadi et al., 2020), as well as with regions of the genome that are LOH.
CN9 is associated with a poor disease specific survival in a pan-cancer analysis, as well as specifically in kidney renal clear cell carcinoma and low grade glioma. CN9 is also associated with an increased leucocyte fraction in low grade glioma and high grade serous ovarian cancer (see Thorsson et al., 2018), and with an increased hypoxia score from gene expression data (see Eustace et al., 2013 & Yang et al., 2018).
CN9 is positively associated with TP53 mutation (SNV/indel) in colorectal carcinoma and uterine/endometrial carcinoma, and with PTEN mutation in low grade glioma. In contrast, CN9 is negatively associated with TP53 mutation in glioblastoma, IDH1 and CIC mutation in low grade glioma and PTEN, CTNNB1, CTCF and ARID1A mutation in uterine/endometrial carcinoma.
CN9 is positively associated with EGFR amplification and CDKN2A loss in glioblastoma, and with CDKN2A loss in low grade glioma.
Additionally, CN9 is recurrently seen around tumour suppressor genes e.g. CDKN2A, RB1, TP53 and STK11, in combination with other focal LOH signatures.
Acceptance criteria
| Background | Identification study | First included in COSMIC | |
|---|---|---|---|
| Steele et al. 2022 Nature | v3.3 | ||
| Identification | NGS technique | Different variant callers | Multiple sequencing centres |
| SNP6 | Yes | No | |
| Technical validation | Validated in orthogonal techniques | Replicated in additional studies | Extended context enrichment |
| No | No | - | |
| Proposed aetiology | Mutational process | Support | |
| Chromsomal instability | Simulation | ||
| Experimental validation | Experimental study | Species | |
| - | - | ||
Summary of the technical and experimental evidence available in the scientific literature regarding the validation of the mutational signature.
Tissue distribution
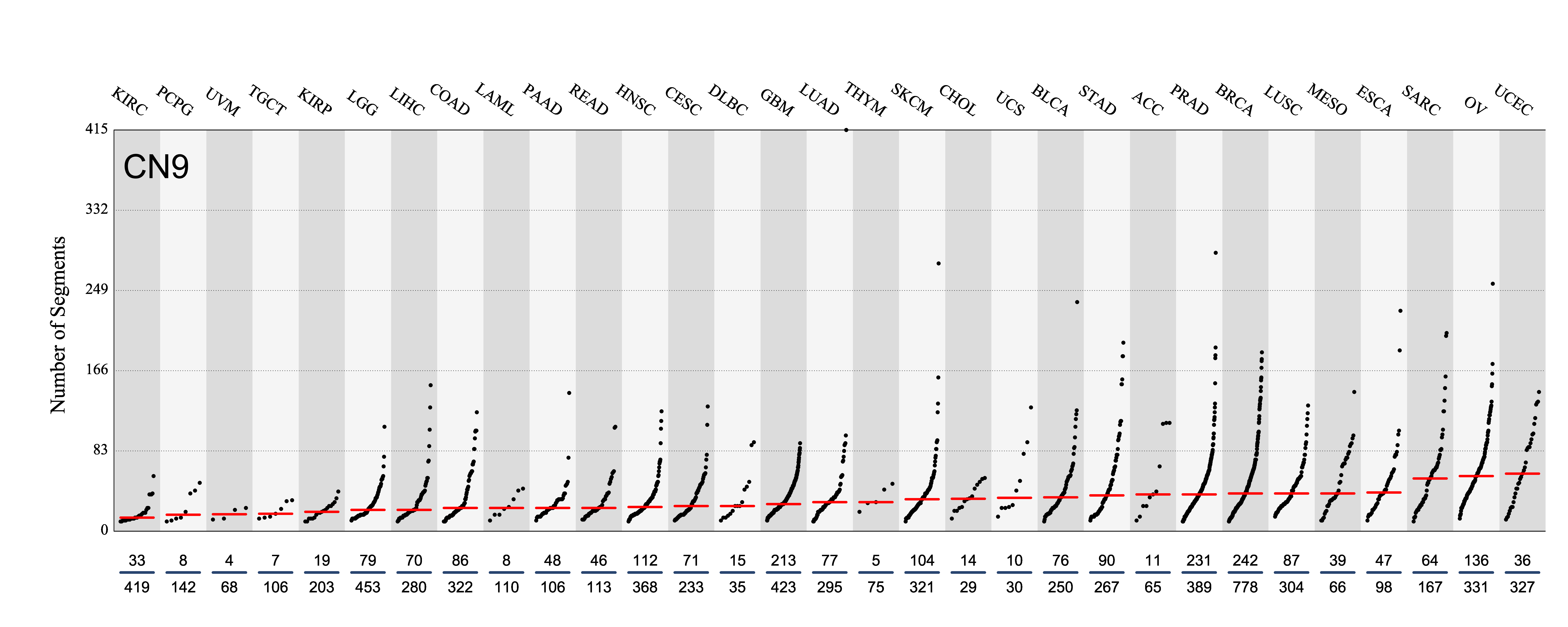
Number of segments attributed to the copy number signature across the cancer types in which it was found. Each dot represents an individual sample and only samples where the signature is found are shown.
The numbers below the dots for each cancer type indicate the number of high confidence tumours in which at least 10 mutations were attributed to the signature (above the blue horizontal line) and the total number of high confidence tumours analysed (below the blue horizontal line). Only high confidence data are displayed: samples with reconstruction accuracy >0.90. Note that due to the nature of copy number data, a sample with e.g. 22 counts of CN1 (a majority diploid unaltered signature, with large segment sizes) may span the whole genome, while a sample with e.g. 70 counts of CN8 (a highly segmented chromothripsis amplification signature, with small segment sizes) may span a small region of the genome.